23
Jun
They are usually located in the ventricles, often near the foramen of monro, where they can cause an obstruction if they grow too large, leading to increased intracranial pressure. They are rare and achievements in their management are often the result of small series and case reports.
Subependymal giant cell astrocytomas. Hindawi�s academic journals cover a wide range of disciplines. It is one of the intracranial lesions found in tuberous sclerosis complex (which include subependymal nodules, cortical tubers, retinal astrocytoma and subependymal giant cell astrocytoma), but cases without such. Targeted therapy with everolimus or sirolimus may be used instead of surgery, to shrink the tumors.
They are rare and achievements in their management are often the result of small series and case reports. Serial neuroimaging demonstrates a continuum. The aim of this study was to determine whether they could be differentiated during childhood and at an early preclinical stage, from subependymal nodules without any growing potential.
Tuberous sclerosis complex is a genetic disorder characterized by benign tumor growth including lesions in the ventricular system of the brain known as subependymal giant cell astrocytomas. Subependymal giant cell astrocytoma is a rare, benign brain tumor that arises from astrocytes, the supportive cells in the nervous system. Intraventricular astrocytomas (subependymal giant cell astrocytomas) of tuberous sclerosis have a poor prognosis due to the obstruction of csf flow.
Ad stroke research and treatment invites cerebral circulation & associated disease research. Subependymal giant cell astrocytoma is a rare tumor that occurs in the wall of the lateral ventricle and foramen of monro and, rarely, in the third ventricle. To date, no radiographic features have been identified that will accurately predict which subependymal nodules will grow and require treatment.
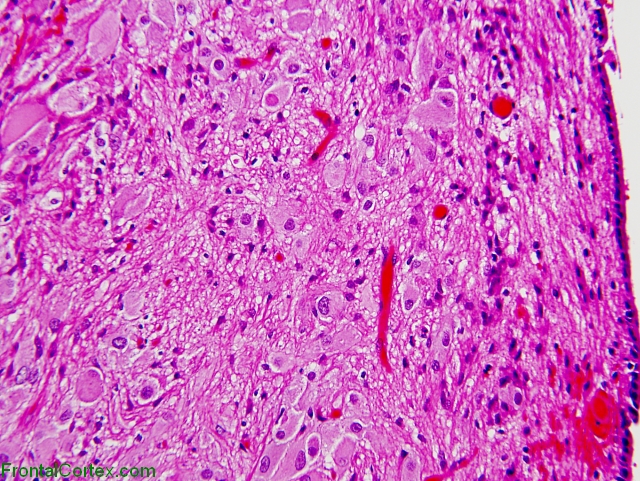
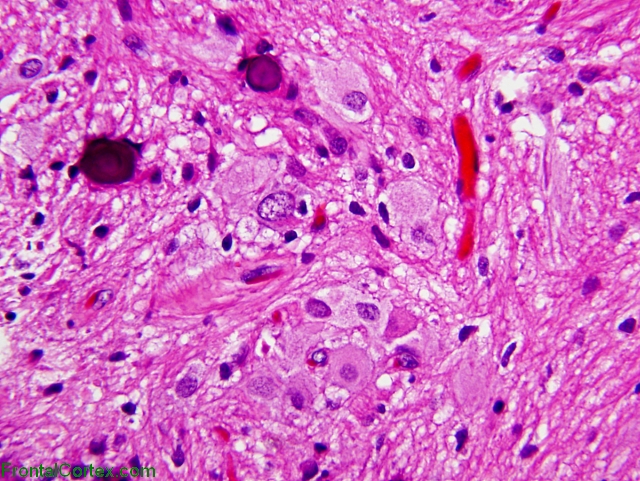
They are usually located in the ventricles, often near the foramen of monro, where they can cause an obstruction if they grow too large, leading to increased intracranial pressure. An alternative may be the use of everolimus, which inhibits the mammalian target of rapamycin, a protein regulated by gene products involved in the tuberous sclerosis complex. As the name implies, these tumors are composed of large ganglioid astrocytes, which are located along the wall of the lateral ventricle.
Purpose of review subependymal giant cell astrocytomas (segas) are grade i astrocytic tumors according to who 2016, mainly associated to tuberous sclerosis complex. They often cause hydrocephalus and are potentially accessible to a surgical treatment. Subependymal giant cell astrocytomas are one of the three major intracranial lesions found in tuberous sclerosis complex.
Subependymal giant cell astrocytomas are characteristic brain tumors that occur in 10% to 20% of tuberous sclerosis complex patients and are almost exclusively related to tuberous sclerosis complex. This analysis focuses on the clinical presentation, management, and associated burden of subependymal giant cell astrocytomas in patients with tuberous sclerosis complex. Our aim is to evaluate morbidity and results after surgery in symptomatic and asymptomatic patients.
The main purpose of this review was to summarize recent developments in diagnosis. Though histologically benign, segas can lead to serious neurological complications, including hydrocephalus, intractable seizures and death. Subependymal giant cell astrocytoma (sega) is a slowly growing tumor of unknown histogenesis mainly arising in the periventricular regions adjacent to the foramen of monro [1, 2], which causes increased intracranial pressure, seizures, and focal neurologic signs.
Benign, slowly growing tumor typically arising in wall of lateral ventricles and composed of large ganglioid astrocytes. Subependymal giant cell astrocytomas usually grow slowly, but their progression ultimately leads to the occlusion of the foramen of monro, with. Though histologically benign, they can cause serious neurologic symptoms, leading to death if untreated.
Usually associated with tuberous sclerosis, an autosomal dominant syndrome due to mutations in tsc1 gene on #9q34 (hamartin protein) and tsc2 gene on #16p13.3 (tuberin protein) who grade i.
Previous post
Sunscreen recommended by dermatologistsNext post
Sub q test injections